Madrid, ESP. Un equipo de investigadores del Consejo Superior de Investigaciones Científicas (CSIC) y la Universidad Autónoma de Madrid identificó una proteína que podría ayudar a proteger las neuronas en personas con enfermedad de Huntington, un padecimiento neurodegenerativo con escasas opciones terapéuticas.
Al activar esta proteína, denominada PKD1, en una zona específica del cerebro, los científicos lograron que las neuronas resistieran mejor el daño que causa su destrucción en esta enfermedad. El hallazgo, publicado en la revista Cell Death & Disease, podría abrir nuevas vías para desarrollar tratamientos que frenen el avance de este padecimiento.
La investigación, dirigida por Teresa Iglesias, fue realizada por el Instituto de Investigaciones Biomédicas Sols-Morreale (IIBM-CSIC-UAM) en colaboración con el equipo de José J. Lucas, y con la participación de los grupos de Eva Porlan y Miguel R. Campanero, estos tres últimos del Centro de Biología Molecular Severo Ochoa (CBM-CSIC-UAM).
Características de la enfermedad de Huntington
La enfermedad de Huntington, descrita por primera vez en 1872 por el médico estadunidense George Huntington, es un trastorno hereditario que provoca la degeneración progresiva de las neuronas. Está causada por una mutación en el gen HTT, que origina una secuencia anormalmente larga de repeticiones y da lugar a una versión defectuosa de la proteína huntingtina.
En condiciones normales, esta proteína cumple funciones esenciales durante el desarrollo embrionario y a lo largo de la vida en el cerebro, pero su versión alterada se acumula en las neuronas y las deteriora de forma progresiva.
Aunque la mutación está presente desde el nacimiento, los síntomas suelen aparecer entre los 30 y los 50 años. El daño en el cerebro comienza en una zona llamada estriado, que controla los movimientos voluntarios y funciones cognitivas. Es allí donde las neuronas comienzan a morir en las primeras fases de la enfermedad, mucho antes de que se manifiesten los primeros síntomas.
Ralentizar la neurodegeneración con PKD1
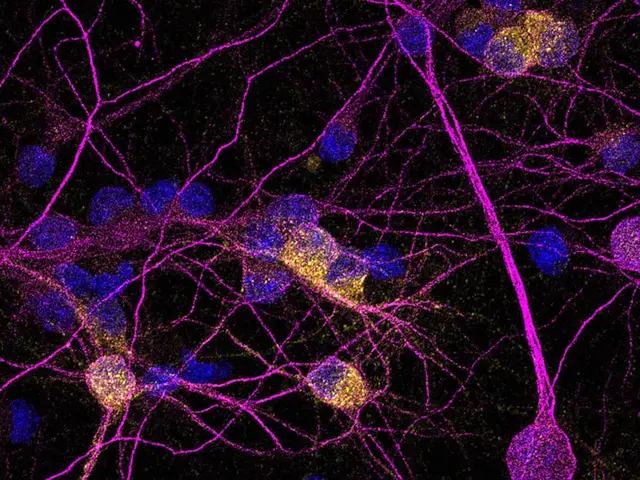
El nuevo estudio se centra en la proteína PKD1, que ayuda a las neuronas a defenderse del estrés oxidativo, un tipo de daño celular causado por exceso de actividad química en el cerebro. Este estrés puede ser especialmente destructivo cuando se combina con la excitotoxicidad: una sobreestimulación de las neuronas que, en vez de facilitar la comunicación celular, termina por dañarlas.
Los investigadores descubrieron que, en pacientes con Huntington, la cantidad de PKD1 está reducida en las neuronas del estriado, lo que las hace más vulnerables al daño. En contraste, en otras células cerebrales, como los astrocitos, esta proteína aparece aumentada, lo que sugiere un desequilibrio en su regulación según el tipo celular.
El avance clave del estudio fue el uso de una herramienta molecular diseñada específicamente para activar PKD1 solo en las neuronas. Al aplicarla en cultivos neuronales y en el cerebro de ratones con un modelo de Huntington, observaron que las neuronas tratadas resistían mucho mejor el daño causado por la excitotoxicidad.
“La actividad de esta proteína quinasa es clave para la supervivencia neuronal. Su pérdida contribuye al deterioro temprano en Huntington, pero su potenciación podría ralentizar la neurodegeneración”, explicó Ana Simón, una de las investigadoras del proyecto.
Implicaciones terapéuticas y contexto internacional
Aunque desde el CSIC recalcan que se trata aún de una investigación en fase experimental, aseguran que el descubrimiento representa un paso importante hacia nuevas terapias para la enfermedad de Huntington. La estrategia de activar PKD1 podría aplicarse en el futuro mediante técnicas de terapia génica o el uso de moléculas que imiten su función protectora.
“Descubrir los mecanismos moleculares que PKD1 pone en marcha para proteger a las neuronas podría tener un gran impacto también en otras enfermedades neurológicas”, destacó Álvaro Sebastián, otro de los autores del estudio, quien ahora lidera su propio grupo en la Universidad Complutense de Madrid.
Este estudio forma parte de la colaboración entre los equipos mencionados, en el marco del Centro de Investigación Biomédica en Red (Ciber, ISCIII), dentro de las áreas de Enfermedades Neurodegenerativas (Ciberned) y Cardiovasculares (Cibercv).
Notas adicionales sobre prevalencia y proyección clínica
La enfermedad de Huntington afecta a aproximadamente 5 a 10 personas por cada 100 mil habitantes en Europa y América del Norte, pero es más rara en Asia y África. En México, aunque no existen cifras oficiales precisas, estimaciones clínicas indican una prevalencia cercana a 1 por cada 100 mil personas. Actualmente no existe cura para esta enfermedad, y los tratamientos disponibles se limitan al manejo sintomático.
La proteína PKD1 (Protein Kinase D1) pertenece a una familia de quinasas serina/treonina implicadas en procesos clave como la supervivencia celular, el tráfico vesicular y la respuesta al estrés oxidativo. Su función neuroprotectora ya había sido explorada en otros modelos de daño cerebral, como el ictus y la enfermedad de Alzheimer, lo que refuerza su potencial como diana terapéutica común en distintas patologías neurodegenerativas.
Según datos del Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, en México los esfuerzos de investigación en enfermedades neurodegenerativas han crecido en la última década, pero siguen siendo limitados por la falta de financiamiento sostenido. El modelo utilizado en este estudio con ratones transgénicos para Huntington es uno de los estándares internacionales en el desarrollo preclínico de terapias.
La revista Cell Death & Disease, donde fue publicado el estudio, pertenece al grupo Nature Publishing Group y cuenta con un factor de impacto superior a 9, lo que posiciona a este trabajo entre la investigación de alta calidad científica en el ámbito de la biomedicina traslacional.
En paralelo a estos avances en España, otros centros internacionales, como el CHDI Foundation en Estados Unidos, también exploran estrategias de neuroprotección en Huntington, incluyendo inhibidores de agregación de huntingtina mutada, edición genética con CRISPR/Cas9 y antisense oligonucleotides (ASO). La activación de PKD1 podría, eventualmente, combinarse con algunas de estas aproximaciones para potenciar su eficacia clínica.
Con información de Excelsior.

